Research Information
Program package of ELSES (Extra Large Scale Electronic Structure Calculation) is designed for large-scale atomistic simulation with quantum mechanical freedom of electrons manipulating a large Hamiltonian matrix. Program package of ELSES consists of two parts; one is the standard molecular dynamics simulation program and the other is the electronic structure calculation program. The latter is the unique part of ELSES and enables us to calculate the electronic structures of extra-large scale systems.
The electronic structure calculation in ELSES is based on the tight-binding formalism of local basis set of wave-functions (the locality of Hamiltonian) and density matrix. Further, we have developed a set of solver methods for large-scale linear equations without calculating eigen-states in a fully quantum mechanical description of electron systems. Among them, the subspace diagonalization method and the shifted Conjugate Orthogonal Conjugate Gradient method are based on the Krylov subspace. These two solver methods are essentially important and are the key solver methods in ELSES, which makes the computational load, both CPU time and memory size, proportional to the system size.
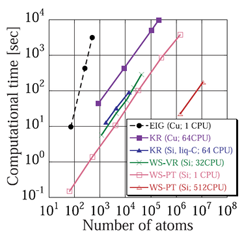
The figure below shows the Order-N property of ELSES, the linear dependence of the CPU time on the number of atoms in the system. The test calculation has been done both in semiconductors and metals and the largest systems we have tested is of 10–7 atoms.
Target of our development
ELSES enables us to achieve MD simulations with electronic and atomistic structural changes in extra-large scale systems of 100 nm size, and possibly in process of nano-devices required in industries. However, at the present stage, ELSES needs further efforts of development for large varieties of chemical elements and compounds, and the usability of the software, and also experience of applications to variety of conditions such as surfaces, interfaces, defects, liquids and also metallic systems.
Future program of further development
Development of general tight-binding formalism applicable to large variety of compounds
Treatment of self-consistent charge
Further efficient computation algorithm in electronic structure calculation
Current and conductivity analysis with finite bias voltage
Further efficient usability in analyzing electronic and atomistic structures